


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
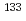 CsI +
CsI + 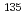 Cs
Cs  Cs + ICs
Cs + ICs小林 孝徳*; 松岡 雷士*; 横山 啓一
Computational and Theoretical Chemistry, 1150, p.40 - 48, 2019/02
被引用回数:1 パーセンタイル:1.99(Chemistry, Physical)セシウムの同位体分離法の開発に関連して、ヨウ化セシウム分子とセシウム原子の衝突によるセシウム交換反応の反応速度定数を量子化学計算及び擬古典トラジェクトリー計算により評価した。その結果、3.6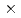 10
10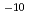 cm
cm /molecule/sという大きな値が得られた。また、わずかながら正の温度依存性を持つことが示され、長距離相互作用による引力ポテンシャルと解離プロセスの影響がその原因と考えられた。
/molecule/sという大きな値が得られた。また、わずかながら正の温度依存性を持つことが示され、長距離相互作用による引力ポテンシャルと解離プロセスの影響がその原因と考えられた。
CsI (v = 0, j = 0) + Cs Cs + ICs小林 孝徳*; 松岡 雷士*; 横山 啓一
日本エネルギー学会誌, 96(10), p.441 - 444, 2017/10
セシウム交換反応CsI (v=0, j=0) + Cs Cs + ICsの反応断面積を調べるため、ab initio分子軌道法計算により作成したポテンシャルエネルギー面を用いた準古典的トラジェクトリー計算を行った。ポテンシャルエネルギー面から反応中間体Cs Iの生成に入口障壁がないこと、2つのCsI結合が等価であることが明らかになった。トラジェクトリー計算により反応断面積は衝突エネルギーの増加と共に単調増加することが分かった。CsI分子の初期内部状態がv=0, j=0の場合の反応速度定数は500-1200Kの温度範囲で310cm molecule
Iの生成に入口障壁がないこと、2つのCsI結合が等価であることが明らかになった。トラジェクトリー計算により反応断面積は衝突エネルギーの増加と共に単調増加することが分かった。CsI分子の初期内部状態がv=0, j=0の場合の反応速度定数は500-1200Kの温度範囲で310cm molecule s程度と見積もられ、わずかながら負の温度依存性が見られた。
s程度と見積もられ、わずかながら負の温度依存性が見られた。
 D)+CH
D)+CH insertion reaction
insertion reaction高柳 敏幸; 黒崎 譲*
Journal of Molecular Structure; THEOCHEM, 492, p.151 - 158, 1999/00
N(D)+CH反応について分子軌道法を直接用いた古典的トラジェクトリー計算を行った。反応の遷移状態から出発し、反応座標方向にエネルギーを与えて計算を行った。N(D)原子がCHのCH結合に挿入して生成するCH NH中間体ラジカルの寿命は非常に短いことがわかった。この結果は実験結果と定性的に一致する。このことからエネルギーはおもにCHとNHフラグメントの相対運動に分配されると予測することが可能である。
NH中間体ラジカルの寿命は非常に短いことがわかった。この結果は実験結果と定性的に一致する。このことからエネルギーはおもにCHとNHフラグメントの相対運動に分配されると予測することが可能である。
F molecule高柳 敏幸; 黒崎 譲*; 田池川 浩人*
Int. J. Mass Spectrom. Ion Process, 176(3), p.227 - 235, 1998/00
被引用回数:4 パーセンタイル:21.15(Physics, Atomic, Molecular & Chemical)CClF分子のイオン化及び電子付着過程に引き続いて起こる分子のダイナミクスについての情報を得るためにab initioダイナミクス計算を行った。垂直イオン化によって生成するCClFは非常に短い時間でCClFとClに解離する。この時、80%のエネルギーには2つのフラグメントの並進運動エネルギーに変換されることがわかった。一方、垂直電動エネルギーによって生成するCClFアニオンも非常に短い時間内でCClFとCl に解離する。しかしこの場合、エネルギーの大部分はCClFの内部エネルギーに変換する。カチオンとアニオンの解離ダイナミクスの違いについて、ポテンシャルエネルギー曲面の違いを基に議論した。
に解離する。しかしこの場合、エネルギーの大部分はCClFの内部エネルギーに変換する。カチオンとアニオンの解離ダイナミクスの違いについて、ポテンシャルエネルギー曲面の違いを基に議論した。
高柳 敏幸
Bulletin of the Chemical Society of Japan, 68(3), p.764 - 770, 1995/00
被引用回数:0 パーセンタイル:0.01(Chemistry, Multidisciplinary)レーザー誘起反応K+NaCl+h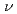 KCl+Na
KCl+Na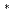 について共線衝突のみを考慮した量子力学的な計算を行った。シュレディンガー方程式はR行列法によって数値的に解いた。量子的な反応確率は反応中間体生成のため多くの共鳴構造をもつ。量子論の結果をホッピングトラジェクトリーの結果と比較した。トラジェクトリー法は反応確率の並進エネルギー依存を正しく与えないが、ラングウ・ツェナー近似は定性的には正しいレーザー波長依存性を示すことがわかった。これらの結果からレーザー誘起反応は、基底状態での反応K+NaClKCl+Naと、基底状態と励起状態間の非断熱遷移の2段階に分けられることがわかった。
について共線衝突のみを考慮した量子力学的な計算を行った。シュレディンガー方程式はR行列法によって数値的に解いた。量子的な反応確率は反応中間体生成のため多くの共鳴構造をもつ。量子論の結果をホッピングトラジェクトリーの結果と比較した。トラジェクトリー法は反応確率の並進エネルギー依存を正しく与えないが、ラングウ・ツェナー近似は定性的には正しいレーザー波長依存性を示すことがわかった。これらの結果からレーザー誘起反応は、基底状態での反応K+NaClKCl+Naと、基底状態と励起状態間の非断熱遷移の2段階に分けられることがわかった。
高柳 敏幸; 横山 淳
Bulletin of the Chemical Society of Japan, 68(8), p.2245 - 2251, 1995/00
被引用回数:17 パーセンタイル:67.07(Chemistry, Multidisciplinary)フッ化ビニルからの4中心HF脱離反応について、古典的トラジェクトリー計算を行った。解析的なポテンシャルエネルギー関数をつくらず、非経験的分子軌道法の計算結果を利用して直接トラジェクトリーの計算を実行した。生成物のエネルギー分布およびベクトル相関の余剰エネルギー依存を計算した。またエネルギー分布の計算結果を実験結果と比較し、定性的に一致することがわかった。より定量的な結果を得るためには、より精度の高いレベルの分子軌道計算が必要となることがわかった。
